Structural analysis of proteins based on deep learning
Biotech Organization | 50 Employees
Machine learning approaches for protein structure prediction, one of the most fundamental problems in bioinformatics and structural biology.
- Modelling proteins at the molecular level involves using amino acid sequences and evolutionary data to predict protein structures.
- Designing proteins to have desired functionality, or predicting their properties or behaviour, is an essential component of understanding and engineering biological systems at the molecular level.
Furthermore, all naturally occurring proteins result from random variants that arise under various selective pressures.
Since predicting protein structure is such a complex task, it is frequently decomposed into four different levels:
- Predicting the structure of amino acids in 1D
- Predicting spatial relationships between amino acids in 2D
- Predicting the tertiary structure of proteins in 3D predicting the quaternary structure of a multiprotein complex in 4D
- Predicting the quaternary structure of a multiprotein complex in 4D
Machine learning techniques based on stacked neural networks utilize deep learning algorithms to frame functions using affine transformations and non-linear activation functions. They often can extract domain-specific features adapted from data that allow them to perform better than traditional methods. Deep Learning has been a notable impact on digital applications such as image classification, speech recognition, and games.
The application of Deep Learning has achieved striking improvements in model accuracy, especially in the "difficult" target category, where comparative modeling is ineffective. For example, in the CASP13 research, it has been demonstrated that neural networks can be trained to learn the complex mapping from amino acid sequence to 3D protein structure and generalize it to unknown cases. In addition, recent advances in deep generative models for the protein design problem have led to several promising approaches.
In the recent past, deep learning networks have achieved remarkable success in computer vision and natural language processing, small molecule representation, transcription factor binding prediction, chromatin effects prediction, and patient outcomes from electronic health records. This is because deep learning is capable of extracting useful features from raw data.
It is interesting to note that local filters in CNNs scan through the input space and search for recurring patterns that are useful for classification performance in the input space. As a result of stacking multiple CNN layers, deep CNNs can hierarchically compose simple local spatial features into complex ones. As we know, biochemical processes occur locally, but they can be aggregated over time and space to form complicated and abstract interactions. CNN's are generally very successful at extracting features from two-dimensional images, which suggests that the concept of convolution can be extended to three dimensions, which can then be applied to proteins represented as three-dimensional "images."
The modern-day operation involves business agility and synchronization with IT.
As Arocom team, we are flexible to adapt the business vision and provide IT support from building, maintaining and running AI/ML Operations, Reimagine workflows and ensuring the business is uninterrupted.
Data (protein structure datasets)
In this study, the data set has been divided into training data set and evaluation data set, with the training set used as a validation data set in the cross-validation method.
In order to represent the alignment of molecules, these protein structures are converted into Coulomb matrices in order to generate an input that is suitable for convolutional neural networks.
- Training set : all the data for protein decoys and the labels associated with the decoys present in the training set
- Evaluation set : all data for proteins and containing the labels associated with the PDB files in the evaluation set
Modelling:
This approach analyzes protein structures by predicting the ten lowest eigenvalues of a coarse-grained model of the protein's fluctuations that is exactly solvable.
The eigenvalues indicate the movement of the atoms in the model, which are then used to derive a number of global properties. As a result, low eigenvalues arise from the small amount of energy required to excite them and the long time required for the molecule to recover back to its stable structure as a result of the self-correlation process.
It is, therefore, quite simple and straightforward to find representations of the most collective and global properties of the model under analysis from its low-energy eigenvalues.
Input data (Transformed protein data):
Coulomb matrices [100,100]
Desired output:
Ten eigenvalues
The architecture of the network:
To model this data, we used convolutional neural networks. Inputs and outputs, as well as the neural network architecture, is shown below. The neural network is built using Keras, as shown in the figure. K-fold cross-validation is used to fine-tune and find the best model.
Convolutional neural network:
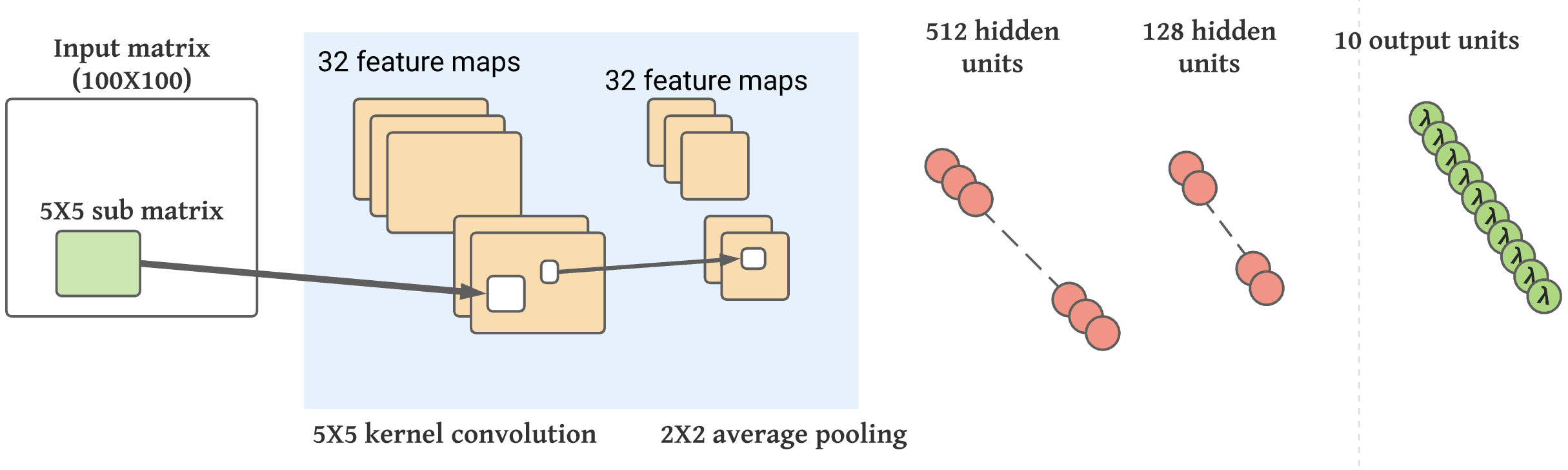
It has been optimized for processing images by restricting the linear transformations of MLP to local convolutions, which compute a pixel state as the linear combination of pixels located next to each other.
Convolution yields just a translated image as a result of translation. In order to reduce the overall scale, convolutions are interspersed with pooling layers that merge blocks of pixels (usually 2 x 2 pixels). In addition to capturing more and more global features, the succession of convolutive and pooling layers will also take into account neighbouring information.
An advantage of this architecture is that the structure of a protein can be represented as a 3D image, making it particularly useful in protein design. Thus, all CNNs developed for processing images-a very dynamic area of artificial intelligence can be applied to protein structure analysis.
Evaluation outputs:
Mean absolute percentage error (MAPE) is used to measure the accuracy of the ten eigenvalues of the model. The models can reach around 73% accuracy in predicting the target values.

Dhaval Mandalia is co-founder of Arocom with 20+ years of experience. He works on various Artificial Intelligence projects in various domains. He enjoys data science, machine learning, data engineering, management and training. He writes blogs about data and management strategies and creates vlogs on various health initiatives. He has been a contributing member on various AI communities. Follow him on LinkedIn & Twitter
.